Cystic Fibrosis
Overview
What is Cystic Fibrosis?
Cystic fibrosis (SIS-tik fye-BROH-sis) is an inherited condition that causes the body to make thick, sticky mucus. The mucus causes problems in the lungs, pancreas, and other organs.
People with cystic fibrosis (CF) get lung infections often. Over time, they can have trouble breathing. They may also have digestive problems that make it hard to gain weight.
Top Things to Know
- Cystic fibrosis (CF) is a condition that causes the body to make thick mucus.
- Mucus can cause problems with breathing and digesting food.
- CF treatments include taking medicine, clearing mucus, and taking enzyme supplements for better digestion.
- Avoiding germs can help prevent the lung infections kids with CF are more likely to get.
What Happens in Cystic Fibrosis?
With CF, there is a change in the gene that makes a protein called cystic fibrosis transmembrane regulator (CFTR). This change causes the body to make abnormal CFTR protein or none at all. Without normal CFTR protein, the cells lining the pathways (tubes) inside some organs make thick, sticky mucus rather than the normal thin, watery kind.
Thick mucus can trap bacteria in the lungs, leading to infection, inflammation, and breathing problems. Mucus also can block the path where digestive enzymes (kinds of proteins) flow between the pancreas and the intestines. This makes it hard for people to digest food and get the vitamins and nutrients they need from it.
Thick mucus can also affect the liver, sweat glands, and reproductive organs.
What Are the Signs & Symptoms of Cystic Fibrosis?
CF can cause symptoms soon after a baby is born. The first sign babies might have CF is a blockage in the intestines called meconium ileus (mi-KO-nee-um IL-ee-us). Other kids don't have symptoms until later on. CF can be mild or severe, depending on the person.
Symptoms of CF include:
- lung infections or pneumonia
- wheezing
- coughing with thick mucus
- bulky, greasy bowel movements
- constipation or diarrhea
- trouble gaining weight or shorter height
- very salty sweat
Some kids and teens also might have:
- nasal polyps (small growths of tissue inside the nose)
- frequent sinus infection
- tiredness
What Causes Cystic Fibrosis?
CF is caused by a change in the CFTR gene. To have CF, a baby must get two copies of the changed CF gene, one from each parent.
How Is Cystic Fibrosis Diagnosed?
Newborn screening tests catch most cases of CF. If the screening test is positive, or if a child has CF symptoms, doctors do a painless sweat test. They collect sweat from an area of skin — usually the forearm — to see how much chloride (a chemical in salt) is in it. People with CF have higher levels of chloride.
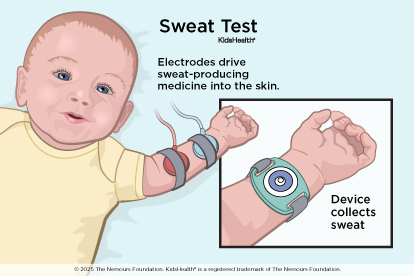
Most children with CF are diagnosed by the time they're 2 years old. But kids with a mild form called atypical CF may not be diagnosed until they’re teens or adults.
Treatment & Home Care
How is Cystic Fibrosis Treated?
People with CF will have it all their lives. Doctors use different medicines depending on a person’s needs. But all people with CF need to:
- Loosen and clear mucus. There are different ways to do this. The doctor might recommend that kids and teens:
- get regular exercise
- use an inhaler or nebulizer
- do breathing exercises and cough on purpose
- wear a therapy vest that shakes the chest
- have chest physical therapy (when a parent or trained person bangs gently on the chest or back)
- Prevent or fight lung infections. People with CF tend to get sick easily. To help prevent infections, wash hands well and often, avoid people who are ill, and stay at least 6 feet away from others with CF since they’re more likely to have and pass on dangerous germs. Taking preventive antibiotics also can help.
- Take enzyme supplements. Most kids with CF need enzymes to help them digest food and get nutrients from it.
- Eat a high-calorie diet and take vitamin supplements, when needed.
How Can Parents Help?
To help your child:
- Follow the treatment plan. Help your child stay as healthy as possible. Give medicines as directed, serve high-calorie meals and snacks, and follow instructions for clearing chest mucus.
- Be encouraging. Help kids find pastimes they enjoy, like art, music, reading, or cooking. It's important for kids with CF to get exercise, so also look for ways your child can stay active. Maybe you can do some of them together.
- Turn to the care team. Your child's care team can offer practical tips on living with CF, and information about clinical trials, support groups, and new therapies.
- Teach self-care as your child gets older. Start early to help your child understand and manage CF. Encourage older kids and teens to handle some parts of their health care, like disinfecting equipment or asking questions at doctor visits. Ask the care team how to help your child get ready for things like going to college or getting a job. Learning about CF and its care helps kids and teens become confident adults managing a chronic health condition.
- Learn all you can about CF. Experts are always working on new treatments to help people with CF have a better quality of life and live longer. Check out resources like the Cystic Fibrosis Foundation website.
Other Common Questions
How Do Doctors Know What Germs Are Causing Cystic Fibrosis Infections?
Kids with CF often get lung and airway infections. A CF sputum respiratory screen or culture helps doctors find and treat germs that cause the infections. Learn about this test.
Can a Spirometry Test Help With Cystic Fibrosis?
Doctors use a spirometry test to check how well the lungs are working. In kids with CF, spirometry can help doctors catch infections and other lung issues early, so they can be treated before they cause more serious problems. Watch a quick video of the breathing test.
What Cystic Fibrosis Tips Can I Give My Child’s Teachers?
It’s useful to teach teachers about CF so they know how to help your child avoid infections and succeed in the classroom. Share this information at school.
 Note: All information is for educational purposes only. For specific medical advice,
diagnoses, and treatment, consult your doctor.
Note: All information is for educational purposes only. For specific medical advice,
diagnoses, and treatment, consult your doctor.
© 1995- The Nemours Foundation. KidsHealth® is a registered trademark of The Nemours Foundation. All rights reserved.
Images sourced by The Nemours Foundation and Getty Images.