Sickle Cell Disease
What Is Sickle Cell Disease?
Sickle cell disease is a group of conditions in which red blood cells are not shaped as they should be. Red blood cells normally look like round discs. But in sickle cell disease, they're shaped like sickles, or crescent moons, instead.
The sickle shaped cells cause problems because:
- They are stiff and sticky and block small blood vessels when they get stuck together. This stops blood from moving as it should, which can lead to pain and organ damage.
- They break down faster than normal red blood cells. That leads to too few red blood cells, a condition called anemia.
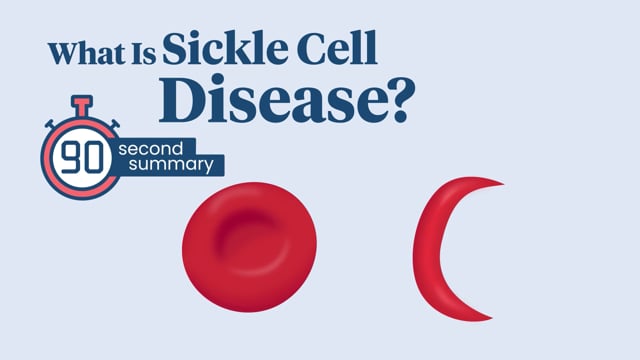
What Is Sickle Cell Disease?
Sickle cell disease causes red blood cells to be curved, or sickle shaped, instead of round. Find out what can happen and how medicine can help.
The two most common symptoms of sickle cell disease are pain and anemia.
The pain caused by sickle cell disease is called a pain crisis or vaso-occlusive crisis. In a pain crisis:
- Pain may happen in any part of the body.
- Cold, stress, illness, or dehydration can bring on pain but often there is not an obvious trigger.
- The pain may last a few hours, a few days, or sometimes longer.
Sometimes pain can be managed at home. But someone with severe pain might need treatment in a hospital.
Signs of anemia include:
- paleness, often seen in the skin, lips, or nailbeds
- tiredness
- dizziness
- being short of breath
- feeling lightheaded
- being irritable
- trouble paying attention
- a fast heartbeat
People with sickle cell anemia may have jaundice (skin and whites of the eyes look yellow). This happens because the sickle-shaped red blood cells break down faster than normal cells.
What Problems Can Happen?
People with sickle cell disease can have problems that need care by a doctor right away, such as:
- Acute chest syndrome: This is caused by inflammation, infection, an blocked small blood vessels of the lung. Signs include chest pain, coughing, trouble breathing, and fever.
- Aplastic crisis: This is when the body temporarily does not make enough red blood cells, which can cause severe anemia. Signs include paleness, extreme tiredness, and a fast heartbeat.
- Hand-foot syndrome: This painful swelling of the fingers and toes (also called ) is the first sign of sickle cell anemia in some infants.
- Infection: Kids with sickle cell disease are at risk for some bacterial infections. It's important to watch for fevers of 101°F (38°C) or higher, which can be signs of an infection. Get medical care right away if a fever happens.
- Priapism: Males with sickle cell disease can have painful, long-lasting erections. If it's not treated quickly, damage can cause problems with getting erections later in life.
- Splenic sequestration crises: The spleen traps the abnormal red blood cells and gets very large. This can lead to a serious, quick drop in the number of red blood cells in the bloodstream. Signs include paleness, weakness or extreme tiredness, an enlarged spleen, and belly pain.
- Stroke: Sickle-shaped cells can block small blood vessels in the brain, causing a stroke. Signs include headache, seizures, weakness in the arms and legs, speech problems, a facial droop, or loss of consciousness.
People with sickle cell disease are also at risk for problems such as leg ulcers, bone or joint damage, gallstones, kidney damage, and eye damage. Kids can have delayed growth and delayed puberty.
The frequency and severity of symptoms and problems varies a lot between different people with sickle cell disease.
What Causes Sickle Cell Disease?
Sickle cell disease is a group of conditions passed down in families through their genes. The type of sickle cell disease a person has depends on the hemoglobin genes each parent passes down to them. Hemoglobin is the protein inside red blood cells that carries oxygen. Someone with sickle cell disease has at least one sickle cell gene. The other hemoglobin gene can be either another sickle cell gene or a gene for a different type of abnormal hemoglobin. The genes cause the body to make hemoglobin that causes the red blood cells to become sickle shaped.
Here’s how sickle cell genes can run in families:
- A child who gets two sickle cell genes, one from each parent, will have sickle cell disease.
- A child who gets a sickle cell gene from one parent and a normal hemoglobin gene from the other parent has sickle cell trait. Most people with sickle cell trait don't have symptoms, but they can pass the sickle cell gene to their children.
- Someone who gets a sickle cell gene from one parent and another kind of abnormal gene from the other parent may have a different form of sickle cell disease, such as hemoglobin SC disease or sickle beta thalassemia.
How Is Sickle Cell Disease Diagnosed?
Sickle cell disease and sickle cell trait usually are found at birth with a blood test during routine newborn screening tests. A second blood test (called a hemoglobin electrophoresis) will confirm the diagnosis.
Sickle cell disease also might be diagnosed before a baby is born with a test on the amniotic fluid or with a sample of tissue from the placenta.
How Is Sickle Cell Disease Treated?
Sickle cell disease is a lifelong condition. Treatment helps people with sickle cell disease avoid problems and stay active. A treatment plan includes:
- Immunizations and daily doses of penicillin to help prevent infection. Kids with sickle cell disease should get all recommended vaccinations, including the pneumococcal, flu, coronavirus (COVID-19), and meningococcal vaccines.
- Folic acid supplements, which can help kids make new red blood cells.
- Medicines to help manage pain when it does happen.
A doctor may recommend other treatments for a child with sickle cell disease, such as:
- hydroxyurea, a daily medicine that makes the cells less sticky. This helps decrease the frequency and intensity of painful episodes and many other problems. It is strongly recommended for many children with sickle cell disease.
- blood transfusions for severe anemia or to treat or prevent some problems
- voxelator (Oxbryta), a daily medicine to reduce sickling and increase the number of red blood cells by helping them hold on more tightly to oxygen
- crizanlizumab (Adakveo), a medicine given by IV infusion that can help make red blood cells more slippery and can reduce pain crises
- L-glutamine, a daily medicine taken by mouth to reduce pain
Stem cell transplant (also called bone marrow transplant) is the only proven cure for sickle cell disease. Transplants are complex and risky but often very successful. They're currently an option only for some patients.
Scientists and doctors are using clinical trials to develop new medicines to treat and prevent problems. They're also studying gene therapy as a potential cure for sickle cell anemia by changing or replacing the abnormal gene that causes it.
When Should I Call the Doctor?
Get emergency medical care right away if your child has any of these problems:
- fever of 101°F (38°C) or higher
- pain that isn't getting better with medicine
- chest pain
- severe headaches or dizziness
- severe stomach pain or swelling
- shortness of breath or trouble breathing
- extreme tiredness
- skin that's yellow or very pale
- a penile erection that is not going away or is painful
- sudden change in vision
- seizures
- weakness or trouble moving part of the body
- slurred speech
- loss of consciousness (passing out)
- numbness or tingling
How Can Parents Help?
When your child has sickle cell disease there is a lot you can do to help:
Learn all you can about sickle cell disease. Tell all caregivers about the condition, how to care for your child, and signs of trouble to watch for.
Take your child to all visits with their doctors and specialists. Keep track of any symptoms and share your concerns.
Help your child avoid pain crisis triggers:
- Encourage your child to drink lots of liquids and get enough rest.
- In cold weather, your child should dress warmly and not stay out too long.
- In hot weather, your child should limit time outdoors and drink lots of fluids.
- Help your child learn ways to manage stress.
- Talk to the doctor about which activities are OK for your child and which to avoid.
- Make sure your child takes all prescribed medicines.
Make healthy choices. As a family, eat healthy foods and stay active. As your child gets older, make sure they know not to smoke, drink alcohol, or use drugs because these can cause pain and other problems.
You also can find more information and support online at:
 Note: All information is for educational purposes only. For specific medical advice,
diagnoses, and treatment, consult your doctor.
Note: All information is for educational purposes only. For specific medical advice,
diagnoses, and treatment, consult your doctor.
© 1995- The Nemours Foundation. KidsHealth® is a registered trademark of The Nemours Foundation. All rights reserved.
Images sourced by The Nemours Foundation and Getty Images.